Click here to download the full course documentation conatining this tutorial.
Mycobacterium tuberculosis is the etiological agent of tuberculosis, and probably the most successful pathogen in human history [1]. The pathogen’s natural cycle of infection entails a complex interaction with the human host and distinct metabolic stages [1, 2]. The capacity of metabolic adaptation is intertwined with the pathogen’s capability to also modulate the expression of several genes, a process that can be driven by the regulation and expression of different sigma factors [3, 4]. This practical session covers a partial analysis of the work reported by Singha et al (2023) which used different M. tuberculosis H37Rv mutant strains to study the regulatory effect exerted by sigma factor A (SigA) and B (SigB) 36690275. For this purpose the authors constructed knock-out mutants of both genes, including a conditional mutant of sigA, by cloning this gene under a anhydro-tetracycline (ATc)-inducible promoter in a sigA knock-out genetic background.
Citation:
Singha B, Behera D, Khan MZ, Singh NK et al. The unique N-terminal region of Mycobacterium tuberculosis sigma factor A plays a dominant role in the essential function of this protein. J Biol Chem 2023 Mar;299(3):102933. PMID: 36690275 https://doi.org/10.1016/j.jbc.2023.102933
The data for this practical is available through the NCBI Gene Expression Omnibus (GEO) database under accession GSE197742. The dataset comprises 10 samples, subjected to RNA-Seq by paired-end sequencing, representing a wild-type control strain (H37Rv), a sigB knockout mutant, and conditional sigA knockout mutant under induced (+ATc) and non-induced conditions (-ATc) Pf4-uninfected strains (2-3 biological replicates each, Table 1):
The practical is divided in three exercises, Exercise 1 pertains the mapping and generation of raw transcript count and has been carried out ahead since the computational steps involved are time consuming. Nonetheless, the commands executed are outlined below along with explanatory context. The second exercise will involve the visualization of mapped reads and the third exercise will cover the identification of differentially expressed genes across the conditions tested.
The initial stage of the analytical process of RNA-Seq data for this practical will consist of mapping the reads to a reference genome following a similar approach as the one already covered in the mapping module for this course. As such, this will require an adequate genome reference file. An annotation file covering all genes in this same reference genome will also be needed. We will use the genome of Mycobacterium tuberculosis H37Rv as reference genome, which has already been downloaded in the FASTA format, GenBank format and annotation (GFF3) from NCBI GenBank (accession NC000962). The following files are therefore already present in the course module directory in the Mtb subdirectory. Let’s start!
From the home directory type:
$ cd Module6/Mtb $ ls
Please note for the following files:
• NC000962_3.fasta – reference genome in the fasta format;
• NC000962_3.gb - reference genome in the GenBank format;
• NC000962_3.gff3 – annotation file in the GFF3 format.
You can look inside these files to confirm its format.
In the same directory you will find compressed FastQ files containing the RNA-Seq raw reads. Each is named according to the Run ID in Table 1 and if this is paired-end sequencing, it will have _1.fastq.gz or _2.fastq.gz appended to the Run ID.
Which one do you have? ____________________________
The mapping steps were done using the following commands:
$ bwa mem NC000962_3.fasta SRR18190582_1.fastq.gz SRR18190582_2.fastq.gz | samtools sort --write-index -o SRR18190582.bam - $ bwa mem NC000962_3.fasta SRR18190583_1.fastq.gz SRR18190583_2.fastq.gz | samtools sort --write-index -o SRR18190583.bam - $ bwa mem NC000962_3.fasta SRR18190584_1.fastq.gz SRR18190584_2.fastq.gz | samtools sort --write-index -o SRR18190584.bam - $ bwa mem NC000962_3.fasta SRR18190585_1.fastq.gz SRR18190585_2.fastq.gz | samtools sort --write-index -o SRR18190585.bam - $ bwa mem NC000962_3.fasta SRR18190586_1.fastq.gz SRR18190586_2.fastq.gz | samtools sort --write-index -o SRR18190586.bam - $ bwa mem NC000962_3.fasta SRR18190587_1.fastq.gz SRR18190587_2.fastq.gz | samtools sort --write-index -o SRR18190587.bam - $ bwa mem NC000962_3.fasta SRR18190588_1.fastq.gz SRR18190588_2.fastq.gz | samtools sort --write-index -o SRR18190588.bam - $ bwa mem NC000962_3.fasta SRR18190589_1.fastq.gz SRR18190589_2.fastq.gz | samtools sort --write-index -o SRR18190589.bam - $ bwa mem NC000962_3.fasta SRR18190590_1.fastq.gz SRR18190590_2.fastq.gz | samtools sort --write-index -o SRR18190590.bam - $ bwa mem NC000962_3.fasta SRR18190591_1.fastq.gz SRR18190591_2.fastq.gz | samtools sort --write-index -o SRR18190591.bam -
Each command allows for mapping of the reads using BWA MEM algorithm and pipes it to samtools in order generate a BAM containing all mapped reads and reference. Next you need to index the BAM file by typing:
$ samtools index SRR18190582.bam $ samtools index SRR18190583.bam $ samtools index SRR18190584.bam $ samtools index SRR18190585.bam $ samtools index SRR18190586.bam $ samtools index SRR18190587.bam
At this point you can already visualize your mapping data but we will leave it to the second part of this practical. For now we need to count the number of reads mapped to each feature/gene. Please note that for this stage you will need to know the position, i.e., start, stop and strand of the genes in your gene. This information is not present in the FASTA reference genome you used to map the reads nor you can extract it directly from the BAM files. This is where the GFF file is needed! You will be using HTSeq to count the reads in the genes, specifically its htseq-count script [7]. By typing:
$ htseq-count #or $ htseq-count --help
You will learn a little bit of the different files needed, arguments to this command and output. You can produce the files containing the raw counts by doing the following:
$ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190582.bam NC000962_3.gff3 > SRR18190582_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190583.bam NC000962_3.gff3 > SRR18190583_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190584.bam NC000962_3.gff3 > SRR18190584_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190585.bam NC000962_3.gff3 > SRR18190585_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190586.bam NC000962_3.gff3 > SRR18190586_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190587.bam NC000962_3.gff3 > SRR18190587_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190588.bam NC000962_3.gff3 > SRR18190588_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190589.bam NC000962_3.gff3 > SRR18190589_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190590.bam NC000962_3.gff3 > SRR18190590_count.txt $ htseq-count -f bam -r pos -s reverse -t gene -i Name SRR18190591.bam NC000962_3.gff3 > SRR18190591_count.txt
Can you check why are we using those extra arguments besides the BAM and the GFF file? Also, please note that the output is being redirected to files named *_count.txt files. Look at their content. Does it resemble something like this:
These files contain the raw read counts for each gene and will be used as input for the Differential Gene Expression analysis in Exercise 3. But before that let us visualize the mapped data so that you can understand better what we have done so far and what these counts represent.
Notice that you already have the files with the raw counts! To download all previously created BAM files and index files for this practical session, please go to:
https://ulisboa-my.sharepoint.com/:f:/g/personal/jperdigao_office365_ulisboa_pt/EkBjH_nIesdIlqBXPIXy3_oB4NG7dg4RAksXHik2lODmbQ?e=fyu1w7and download the bam_files_KP.tar.gz file (use command: tar -xvf bam_files_KP.tar.gz to decompress the file). Run this last command in the Mtb sub-directory after downloading the file to this location.
For the next exercise you can visualize the mapped reads in Artemis. Inside the directory containing the data, type:
$ art
This will start Artemis. You must first load your reference genome, which can be done in multiple ways:
i) you can open the NC000962_2.gb file, containing the genome and features in the GenBank file, or;
ii) you can open the NC000962_3.fasta file and then read in the GFF file so that you can see the different feature tracks.
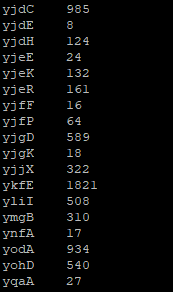
Next, you need to read in your BAM files. You can start by going to File >> Read BAM/VCF and select the first BAM file from the list. Please remember that to load a BAM file in Artemis you need to have the BAM file and the respective .bai index file in the same directory. If everything went ok a BAM window with the mapped reads should appear on top of the genome:
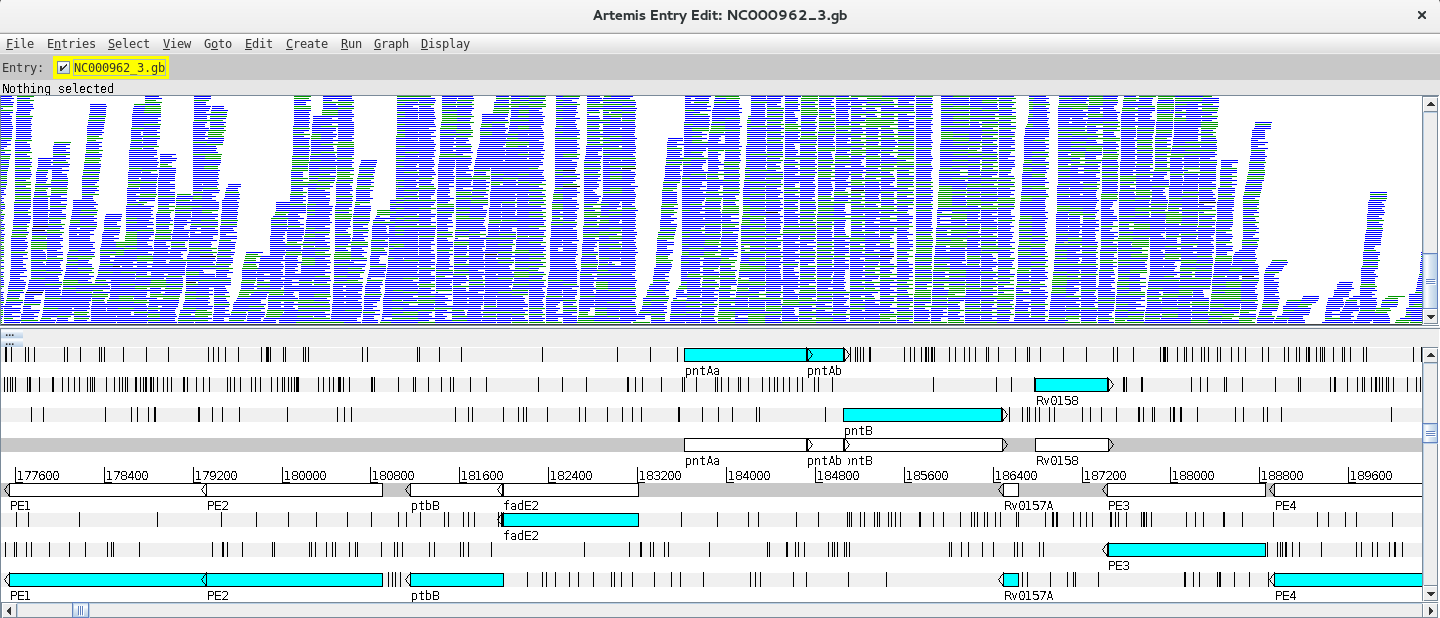
Depending on whether the genome map window is too zoomed in or out you will see the individual reads or a plot. You can change the zoom level in the vertical scroll bar on the right and by right clicking on the BAM window you can also select the type of graph under View (try changing between pileup and coverage for example).
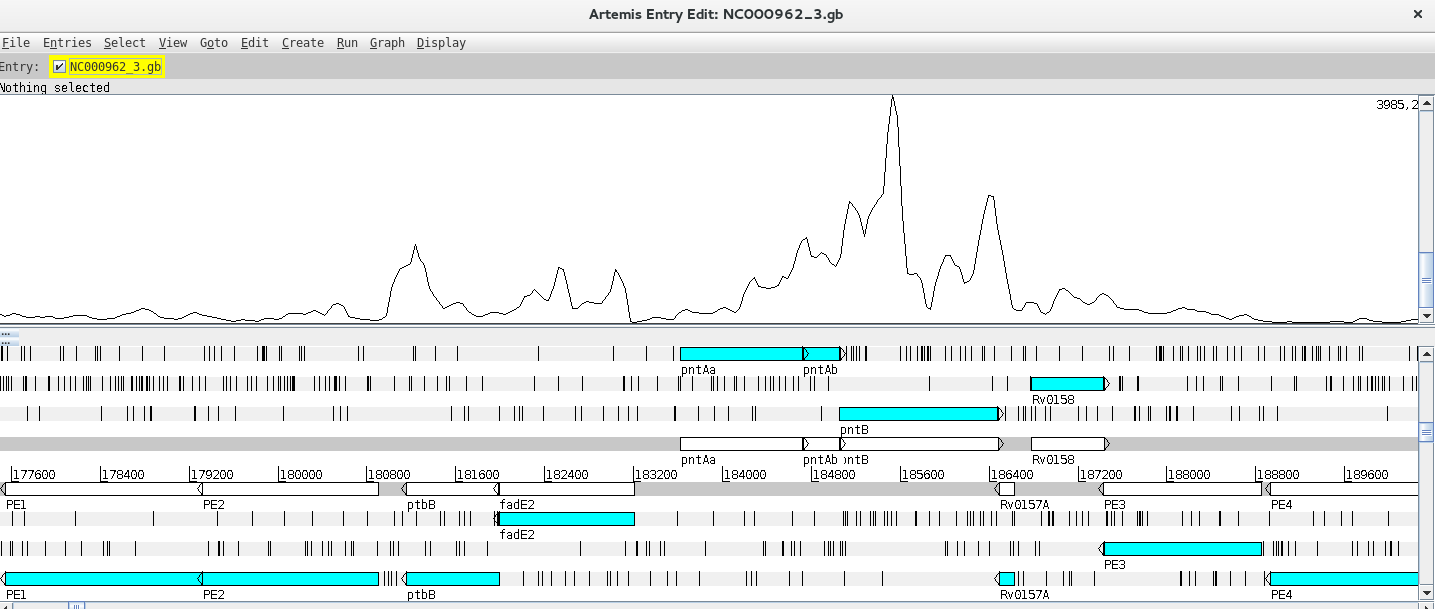
Let’s look at a particular gene, for example fabD12, by using the Navigator (Goto >> Navigator) and typing the gene name in the appropriate search box.
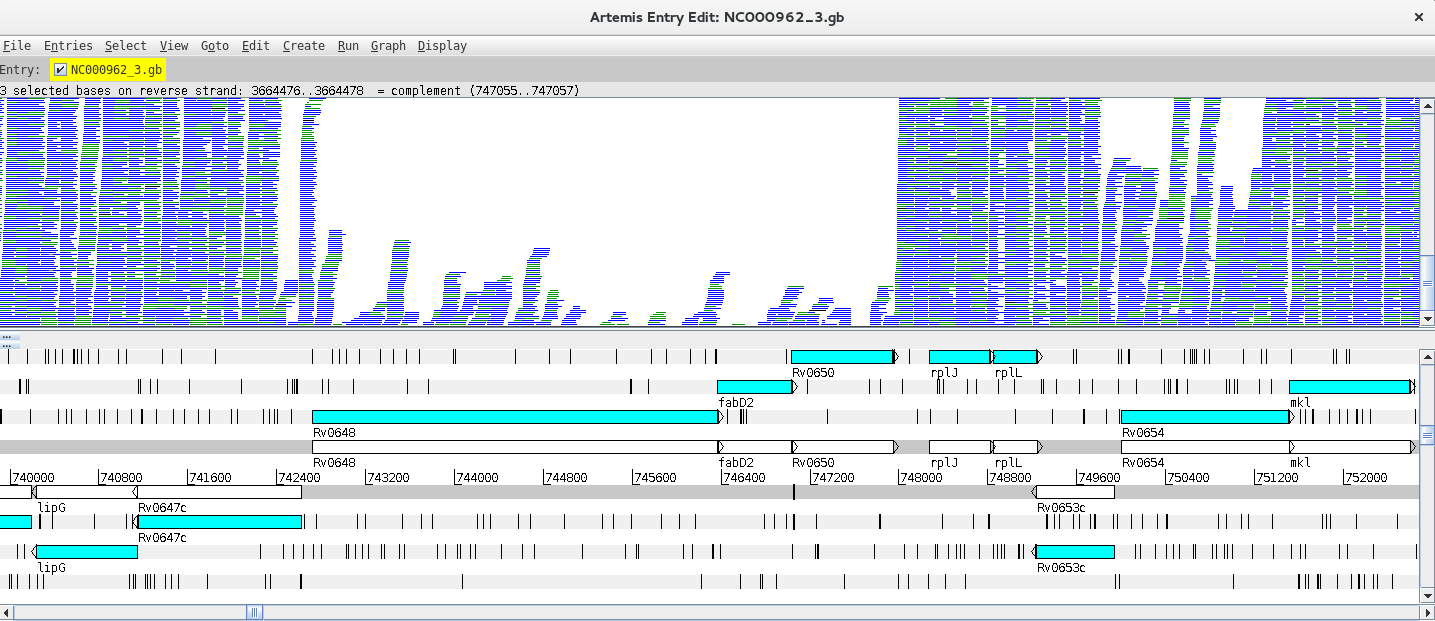
Notice that in the vicinity of these genes, there are some with no/low coverage while others have coverage and there are even reads mapping to intergenic regions. Why is that?
Next, you can add and compare the coverage from other BAM files. To do this just right click on the BAM window, select “Add BAM” and add the different BAM files. Also, changing the Graph type to coverage will facilitate the interpretation of the Data. By right clicking on the BAM window you also have the option to configure these lines (changing the colour, thickness, etc.).
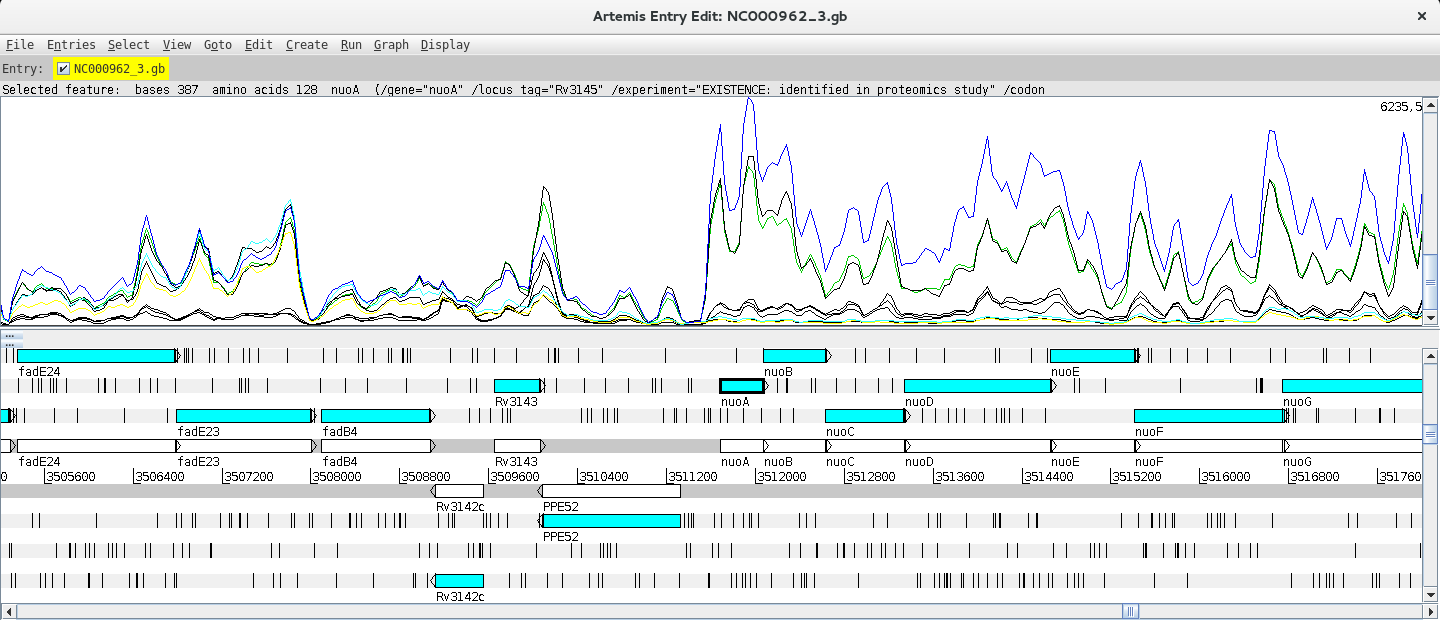
Can you find any differentially expressed genes (DEG) using this approach?
Hint: Try the nuo gene!
This last exercise is aimed at identifying differentially expressed genes (DEGs) by taking the raw counts as input to software that normalizes the number of mapped reads and apply statistical tests to identify DEGs. The most widely used packages for this purpose are the R packages DESeq2 and EdgeR. In this practical we will use DESeq2 to identify DEGs, but code DEG identification using EdgeR is also provided and you can even compare the results between packages [8, 9].
It is also important to stress that using different replicates is a fundamental aspect of DEG analysis as expression levels are variable and depend on several external factors. Biological replicates are therefore essential to account for the variability of biological systems and more accurately compare expression levels between groups/conditions.
The exercise will be carried out in R which involves a different programming language. Let’s go step by step. Let’s start R by typing:
$ R
Inside the R command line we need to load the required libraries for this part:
> library(DESeq2) > library(gplots)
Next we will list the files with counts into an object called sampleFiles and create an object with the condition/genotype. It is important that the order of the genotypes is the same as the order of the listed files:
> sampleFiles<-list.files(pattern="count.txt$")
#Use the same order from the sampleFiles object:
> sampleCondition <- c ("Control","Control","SigBKO","SigBKO",
"SigAKO_ATc","SigAKO_ATc","SigAKO_ATc",
"SigAKO_NoATc","SigAKO_NoATc","SigAKO_NoATc")
We can then create a data frame, which is a type of R object with a table structure called sampleTable:
> sampleTable <- data.frame(
sampleName = gsub("_count.txt","",sampleFiles),
fileName = sampleFiles,
condition = sampleCondition
)
Notice that you defined the columns in this data frame and for sample names you run the object containing the file list through a substitution command to remove the suffix of the file names.
You can check the structure of the data frame just by typing:
> sampleTable
Please check if everything as we will next move to the analytical stage. You can import the HTSeq counts into a DESeqDataSet object and look at the contents of this object by running the following:
> dds <- DESeqDataSetFromHTSeqCount( sampleTable = sampleTable, design = ~ condition ) > dds
Pay attention to the design argument of the first function, you need to specify the grouping factor. This argument can accommodate multiple designs, this is the simplest design scheme in which you specified the name of a single column from your original dataframe.
A pre-filtering step can be carried out to reduce the number of genes with a count below 10. This step is not essential but it can help to reduce the size of the DESeqDataSet object. To do this, run the following:
> keep <- rowSums(counts(dds)) >= 10 > dds <- dds[keep,]
Next, setting the factor levels is important to define the reference level (control group). If this is not specified the levels will be taken in alphabetical order and the reference level will be assumed to be the first one. Below there are two options to set the levels:
> dds$condition <- factor(dds$condition, levels = c("Control", "SigBKO", "SigAKO_ATc", "SigAKO_NoATc"))
# Notice that wt comes first as this is the natural reference group (control)
# or
> dds$condition <- relevel(dds$condition,ref="Control")
The differential expression analysis is carried out using the DESeq function that takes the DESeqDataSet object as input, does the normalization of the counts, estimates dispersion and additional statistical tests. After, the result table can be generated using the results function results:
> dds <- DESeq(dds)
> res <- results(dds, contrast = c("condition", "SigAKO_ATc", "SigAKO_NoATc"))
In the second command we have just used the contrast argument to the results function which might not be necessary but enables you to specifically control the comparison being made across groups. To look at the results table just type:
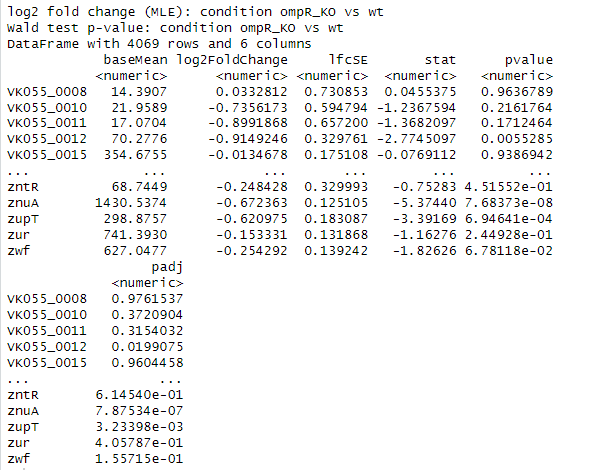
> res
What do you see? Something like this:
You can summarize the results by running:
> summary(res)

You should see the standard adjusted p-value threshold used for the summary and the number of genes with a Log2 Fold Change (LFC) above and below 0.
Next let’s remove results with no adjusted p-values and sort the table by this value:
> res <- res[!is.na(res$padj),] > resOrdered <- res[order(res$padj),] > resOrdered
You can construct a heatmap for the top 50 genes using the following commands:
> counts_heatmap <- counts(dds, normalized = TRUE)
> idx <- rownames(resOrdered)[1:100]
> counts_heatmap <- counts_heatmap[rownames(counts_heatmap) %in% idx,]
> counts_heatmap
> colnames(counts_heatmap) <- c("Control1", "Control2", "SigBKO1", "SigBKO2", "SigAKO_ATc1", "SigAKO_ATc2", "SigAKO_ATc3", "SigAKO_NoATc1", "SigAKO_NoATc2", "SigAKO_NoATc3")
>heatmap.2(as.matrix(counts_heatmap), scale="row", col=greenred(75), Rowv=NA, dendrogram = "col", trace="none", density.info = "none")
Another option is to visualize Gene Plot Counts for multiple genes:
> par(mfrow=c(2,3)) > plotCounts(dds,gene="PE31", intgroup="condition") > plotCounts(dds,gene="PPE60", intgroup="condition") > plotCounts(dds,gene="lpqQ", intgroup="condition") > plotCounts(dds,gene="mutB", intgroup="condition") > plotCounts(dds,gene="Rv3912", intgroup="condition") > plotCounts(dds,gene="Rv0585c", intgroup="condition") > par(mfrow=c(1,1))
You can also do a Principal Component Analysis of the results:
> vsdata<-vst(dds,blind=FALSE) > z <- plotPCA(vsdata,intgroup="condition") > z + coord_fixed(ylim=c(-30,30), xlim=c(-30,30))
Do the global transcriptomic signatures from different conditions cluster in this analysis?
For a Volcano Plot, you can construct a basic one:
> with(res, plot(log2FoldChange, -log10(pvalue), pch=20,main="Volcano plot",xlim=c(-8,8)))
or highlight in blue if the adjusted p-value is below 0.01 or red if the adjusted p-value is below 0.01 and the Log2 Fold Change is higher than 1:
> with(subset(res,padj<.01),points(log2FoldChange, -log10(pvalue),pch=20,col="blue")) > with(subset(res,padj<.01 & abs(log2FoldChange)>2),points(log2FoldChange, -log10(pvalue),pch=20,col="red"))
Moreover, you can write you results table to a csv file, or create individual files of upregulated or downregulated genes based on specific thresholds that you can decide on. For example:
> write.table(resOrdered,file="resOrdered.csv", sep=";",row.names=TRUE, quote = FALSE, col.names=TRUE) ##The following produce different files based on criteria adjusted p-value < 0.05 and Log2 Fold Change > 0.5, sorted by the adjusted p-value: > res_sig<-subset(res,padj<0.05) > res_sig_up<-subset(res_sig, log2FoldChange > 0.5) > res_sig_upOrdered<-res_sig_up[order(res_sig_up$padj),] > write.table(res_sig_upOrdered, file="res_sig_upOrdered.csv", sep=";",row.names=TRUE, quote = FALSE, col.names=TRUE) > res_sig_down<-subset(res_sig, log2FoldChange < -0.5) > res_sig_downOrdered<-res_sig_down[order(res_sig_down$padj),] > write.table(res_sig_downOrdered, file="res_sig_downOrdered.csv", sep=";",row.names=TRUE, quote = FALSE, col.names=TRUE)
You can then open the CSV files in MS Excel or LibreOffice Calc and examine the results. Please note that you must adjust the column headers.
1 Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014; 159: 1497-1509.
2 Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012; 12: 581-591.
3 Sachdeva P, Misra R, Tyagi AK, Singh Y. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. FEBS J. 2010; 277: 605-626.
4 Manganelli R. Sigma Factors: Key Molecules in Mycobacterium tuberculosis Physiology and Virulence. Microbiol Spectr. 2014; 2: MGM2-0007-2013.
5 Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31: 166-169.
6 Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15: 550.
7 Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26: 139-140.
<<-HOME